MP2: Enhanced Multiplex PCR Primer Design for Versatile Applications #
Introduction #
We are pleased to introduce an enhanced version of the multiplex PCR primer design (MPprimer) server, named MP2. This updated version features several improvements. Firstly, the core graph-expanding algorithm has been enhanced to automatically accept or reject-redesign candidate primers, resulting in a more intelligent design process. Secondly, MP2 includes a flexible and comprehensive quality control system, making it suitable for a wide range of applications. These include: 1) designing multiple singleplex primers; 2) creating multiplex primers with excellent cross-species specificity, such as for forensic science; 3) generating primers capable of amplifying both genome A and genome B simultaneously; 4) designing multiplex primers with different amplicon sizes for capillary electrophoresis; 5) producing multiplex primers with similar amplicon sizes for target enrichment, followed by next-generation sequencing. Additionally, the server now includes default settings for important quality parameters, such as avoiding cross-dimers and ensuring that no single nucleotide polymorphisms (SNPs) are located on crucial primer binding sites. Lastly, the server interface has been redesigned for improved usability, with access to more than 100 genomes for quick use. Custom species are also supported upon request.
Availability #
The web server is freely available at https://m4.igenetech.com/mp2/.
Tips #
Whole region design #
To design primers for an entire region, such as human Mitochondrial DNA (mtDNA), the standard BED input should be chrM 0 16569. However, when considering the primer selection at the beginning and end of the region, it is recommended to leave some space. Therefore, the best practice for the entire region is to use chrM 50 16519 as the input, allowing for a 50-base space at both the beginning and end of the region.
Designing Primers for Long-Read Sequencing with Large Product Size #
For instance, when designing primers with a target product size of 5,000 bp, it is recommended to set the “Product Min Size” as 4900 and the “Product Max Size” as 5100. Choosing a smaller range such as 4900-5100 is more efficient in terms of computational resources and memory compared to a broader range like 4000-6000. Setting a wider range could potentially strain computational resources and even necessitate server restarts to prevent memory-related issues in extreme cases.
Web server help #
What is the BED format? #
The BED (Browser Extensible Data) format is a text file format used to store genomic regions as coordinates and associated annotations. We just need the first three columns:
 https://en.wikipedia.org/wiki/BED_(file_format)#Description
https://en.wikipedia.org/wiki/BED_(file_format)#Description
Why we need BED format as targets? Why not just paste sequences? #
In addition to identifying the target sequences, it’s crucial to consider the presence of background DNA—DNA other than the intended targets. When designing primers, each pair is evaluated against the entire genome to minimize the likelihood of non-specific amplification. The BED format plays a key role during this evaluation by distinguishing between target and non-target predicted amplicons. However, differentiating them solely based on input sequences poses a challenge for the algorithm.
How to make the targets in BED format? #
We wrote several blogs:
Why set 50 targets maximum? #
Designing multiplex PCR primers is a meticulous process, involving genome-wide prediction of nonspecific amplicons for each primer pair within a reaction. Typically, the process takes less than 10 minutes for up to 50 targets, with additional time required when incorporating extra background databases. A private link will be created for later access when the task requires a long running time.
To preserve computational resources and prevent misuse, we’ve implemented a cap on the number of targets. Without this restriction, some tasks could run for an extended period (potentially days for thousands of targets), thereby hindering the processing of other users’ tasks.
Researchers with needs exceeding these limits are encouraged to contact the author at quwubin@gmail.com. We are more than willing to accommodate your design tasks efficiently.
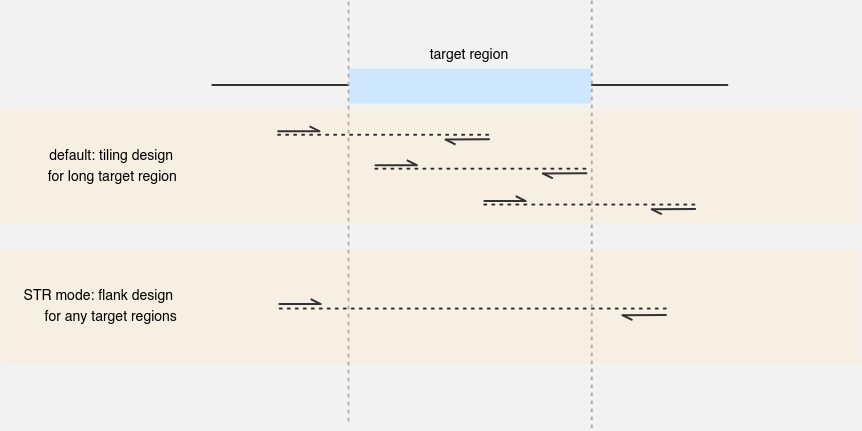
What’s the “STR” mode? #
Short Tandem Repeats (STRs) are regions within the genome consisting of multiple repeating DNA sequences. The STR mode is specifically tailored for designing primers to amplify regions flanking STRs or any target regions intended to be amplified in their entirety.
In scenarios where the target region’s size exceeds the maximum allowed product size, a tiling design strategy is employed with a default setting of two tubes to ensure 100% coverage of the target.
The STR mode is designed to avoid placing primers directly on the target regions, instead selecting them from the surrounding flank areas for optimal amplification.
What’s the target database? And what’s the background databases? How to choose? #
The target database is where the BED-formatted target information originates. Therefore, the chromosome names in the BED format must match those in the genome database.
Background databases are crucial for ensuring that primers do not amplify any unintended sequences from these databases. For instance, in forensic science, it’s essential that a primer pair specifically amplifies the target STR from human genomic DNA, without producing any amplicons from environmental species such as dogs, pigs, or E. coli.
However, the specific needs of researchers can vary across different applications. For example, some researchers might require primers that can simultaneously amplify targets from both genome A and genome B. An example of this is a primer pair designed for human sex identification targeting the AMELX gene:
CCCTGGGCTCTGTAAAGAATAGTG
ATCAGAGCTTAAACTGGGAAGCTG
Result link: https://m4.igenetech.com/spec/52cb28d4-e881-45ff-bf58-8200134c7a98
How to set “Max number of off-target amplicons allowed”? #
In theory, the ideal setting for this parameter is zero, since the goal is to design primer pairs that do not produce non-target amplicons. However, for certain targets, it’s impractical to achieve such specificity. In these cases, setting the parameter to 10 may be necessary. It’s crucial, though, to ensure that there’s a significant base difference between the target and non-target amplicons, allowing them to be distinguished by next-generation sequencing or other analytical methods.
How to set “Amplicon count on background genomes? #
In most cases, it’s advisable to set this parameter to zero. A value greater than zero should be considered based on the specific requirements of the application.
What the “Tube number” means? #
Multiplex PCR is a method that enables the amplification of multiple DNA sequences in a single reaction tube. However, there are situations where multiple tubes may be necessary. The “tube number” refers to how many tubes are required for a given experiment. Here’s how to determine the appropriate tube number:
-
Single Tube for Multiple SNPs: Generally, for amplifying multiple SNPs scattered across the genome, a single tube is sufficient. This is because SNPs are usually well-distributed, allowing for the selection of primers that do not interfere with each other.
-
Multiple Tubes for Large Exons: For targets such as large exons that exceed the maximal product size allowed, a tiling design strategy is employed. This necessitates dividing adjacent primers between two tubes to avoid overlap, meaning the tube number should be set to 2.
-
Batch Singleplex Design: For designing 50 Sanger sequencing primers, the tube number should be set to
Singleplex. This indicates a batch singleplex primer design task, where each primer pair is intended for use in its own reaction. -
Automatic Tube Number Selection: If the optimal tube number is unclear, setting it to
Autoallows the server to automatically determine the best tube number based on the specific requirements of your project.
What’s the “Primer 3’ end SNP-free length”? #
SNPs located within primer binding sites, especially at the 3’ end, can significantly impact the stability of primer-target binding and negatively affect amplification efficiency. Ideally, primers without SNPs are preferred to ensure high amplification efficiency. However, in regions with high variability and numerous SNPs, finding SNP-free primers may be challenging. In such scenarios, one approach is to lower the SNP tolerance parameter to a value like 6 and attempt the primer design process again. Alternatively, using the “STR” mode to amplify the entire highly variable region might be a viable solution.
Currently, SNP databases configured for primer design are available only for the human genome (versions hg39 and hg19) and the mouse genome (mm10). These common SNP databases are sourced from the UCSC website.
What’s the “Validation method”? #
To validate PCR amplicons, commonly employed methods include Sanger sequencing, next-generation sequencing (NGS), and gel electrophoresis. Each validation method may necessitate a distinct design strategy for the primers. Therefore, selecting an appropriate validation method tailored to the specific requirements of your application is crucial before proceeding with primer design.
When to set “Product Min Difference”? #
When the Validation method is set to size discrimination, this configuration aims to design primers that produce amplicons of varied sizes. These size differences enable the discrimination of specific amplicons using size-selective methods such as gel electrophoresis, facilitating the identification and analysis of target sequences based on their length.
What’s “Dimer ΔG”? #
We use this value to filter out the non-significant (those ΔG > this value) dimers.
Waht’s “Hairpin Min Score”? Why hairpin uses score and dimer uses ΔG? #
We utilize this score threshold to eliminate non-significant hairpins, those with scores below this value. In our experience, using a score-based approach for hairpin filtering has proven to be efficient.
What’s “Minimum nonspecific amplicon Tm”? #
This parameter is utilized to eliminate nonspecific amplicons with lower melting temperatures (Tm), as derived from MFEprimer.
What’s “Nonspecific amplicon size range”? #
This range is employed to predict nonspecific amplicons, utilizing parameters from MFEprimer.
Why collopse the “Advanced settings”? #
In the majority of instances, there’s no need to modify these parameter values.
Examples #
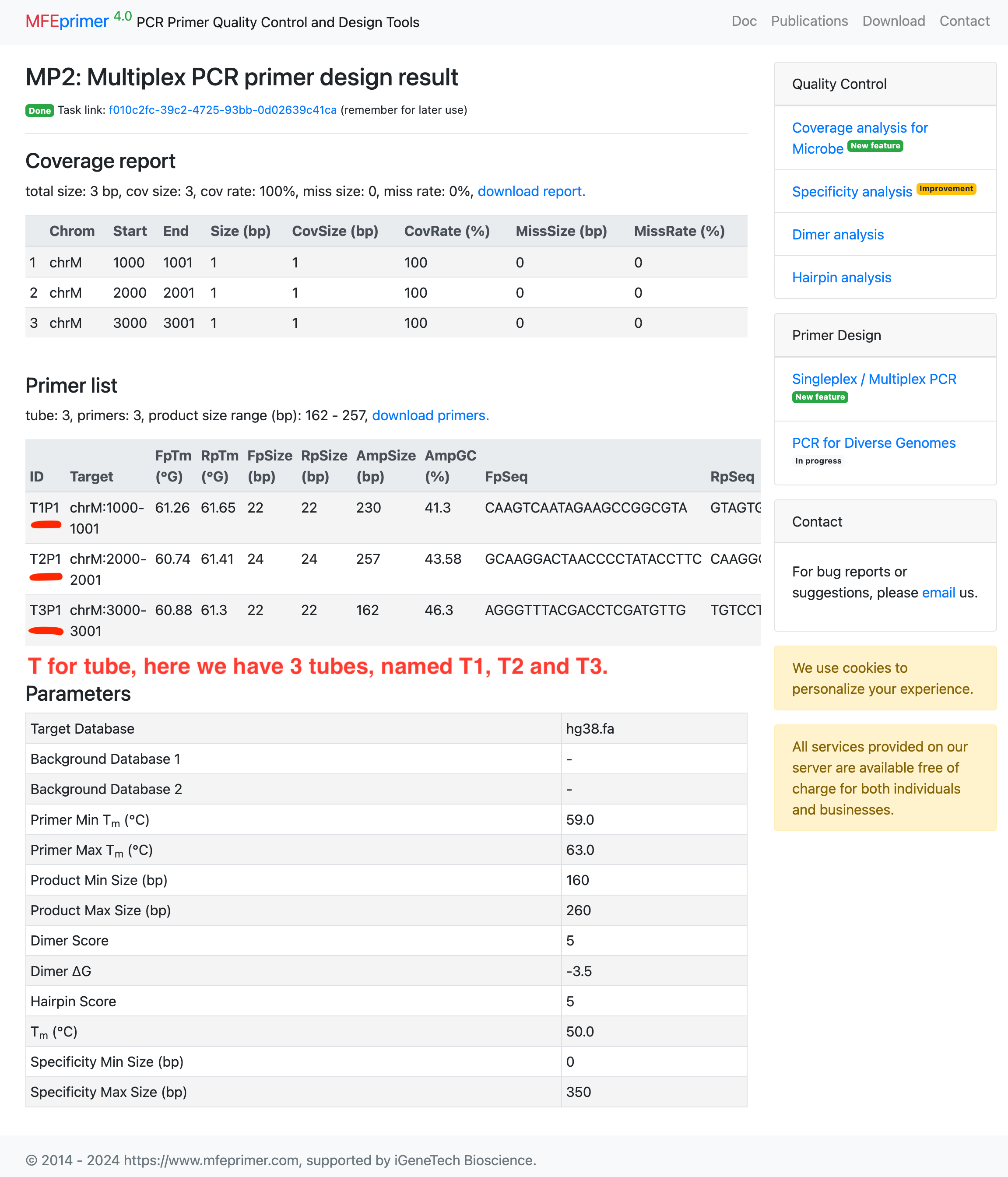
Designing multiple singleplex primers #
- Select the target database;
- Enter the targets in BED format;
- Set “Tube number” = “Singleplex”;.
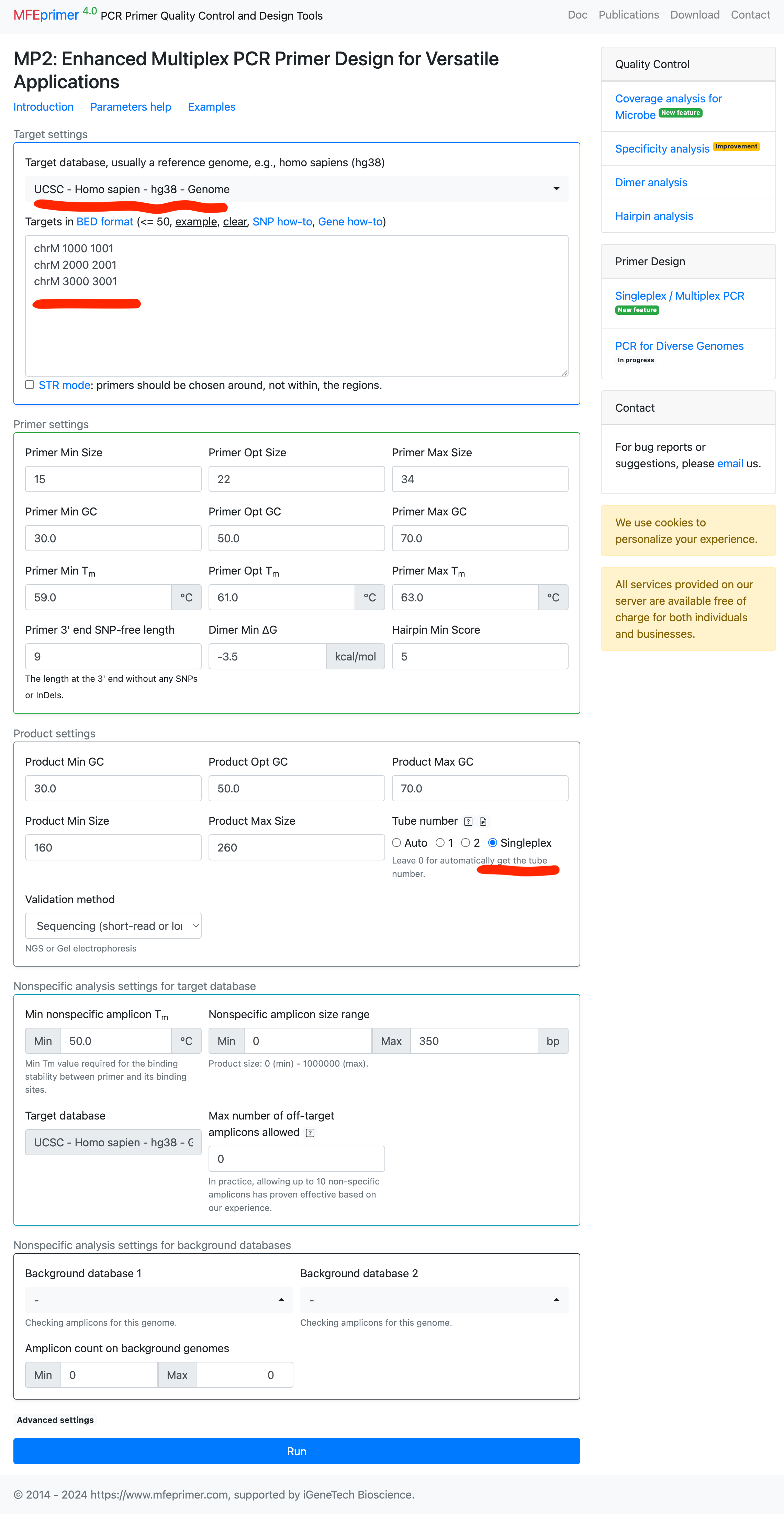
- Below are the results.
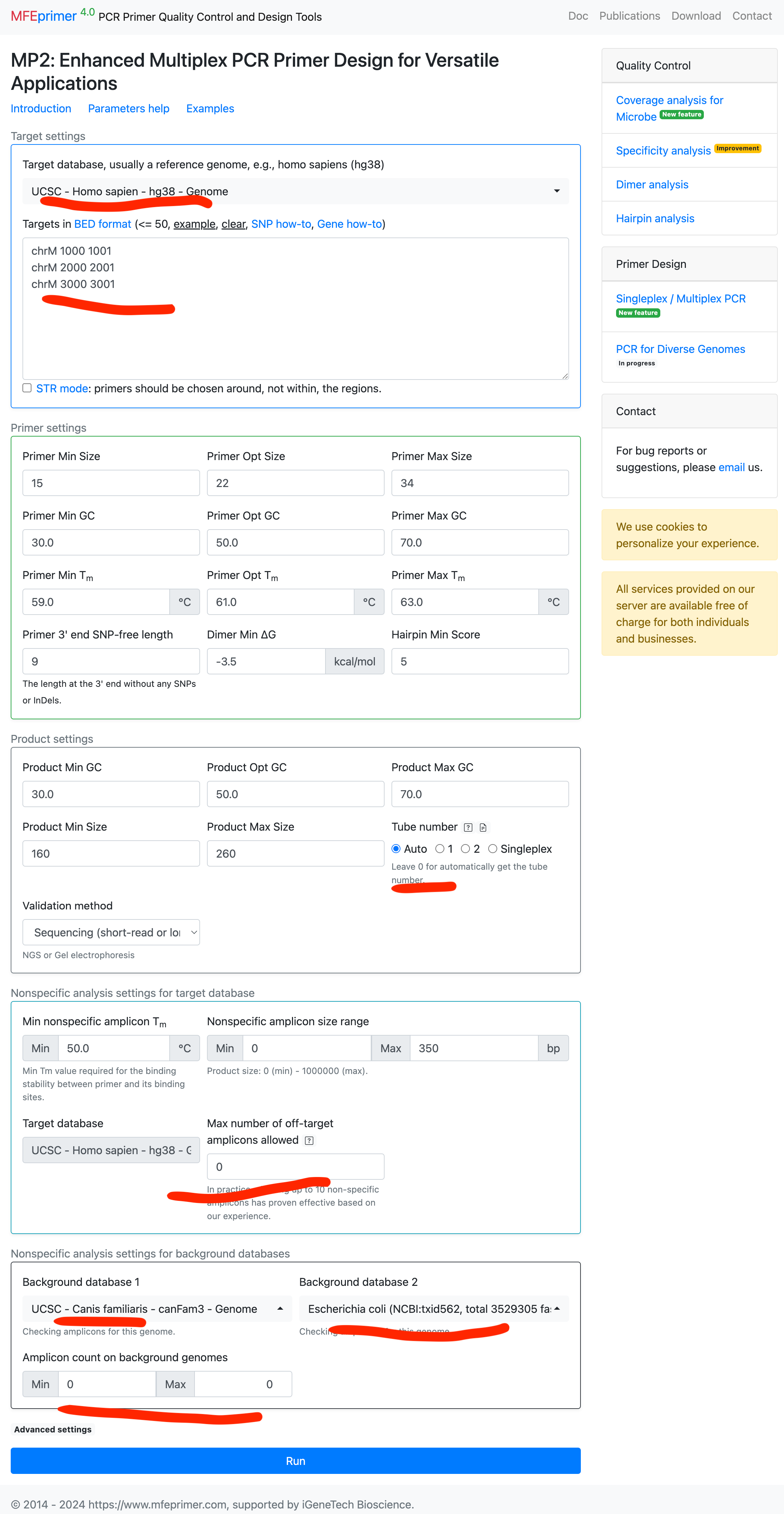
Designing primers with excellent cross-species specificity #
- Select the target database;
- Enter the targets in BED format;
- Also select backgrounds databases;
- And set “Amplicon count on background genomes” = 0;
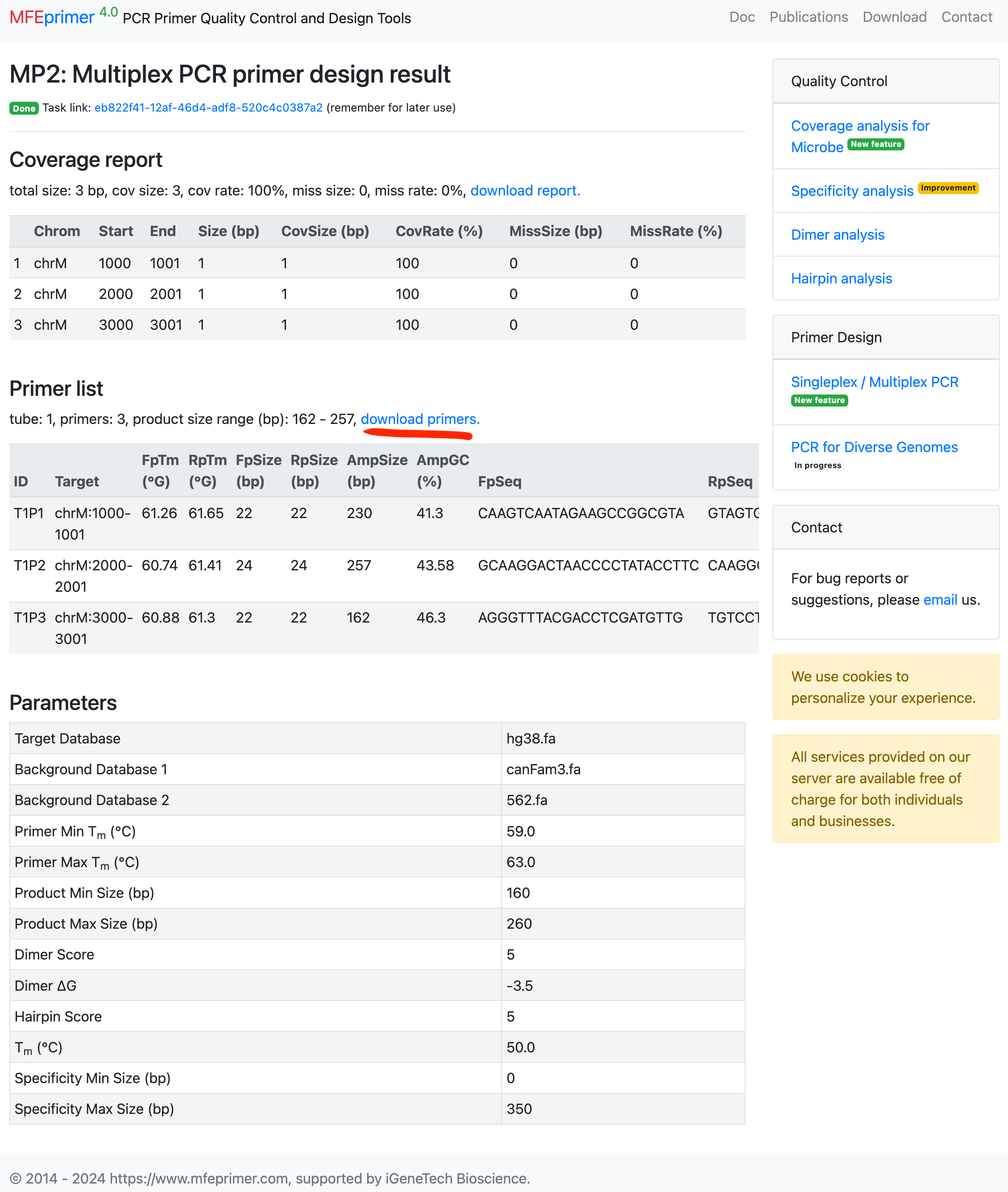
 5. Download the primers and open it in Excel;
5. Download the primers and open it in Excel;
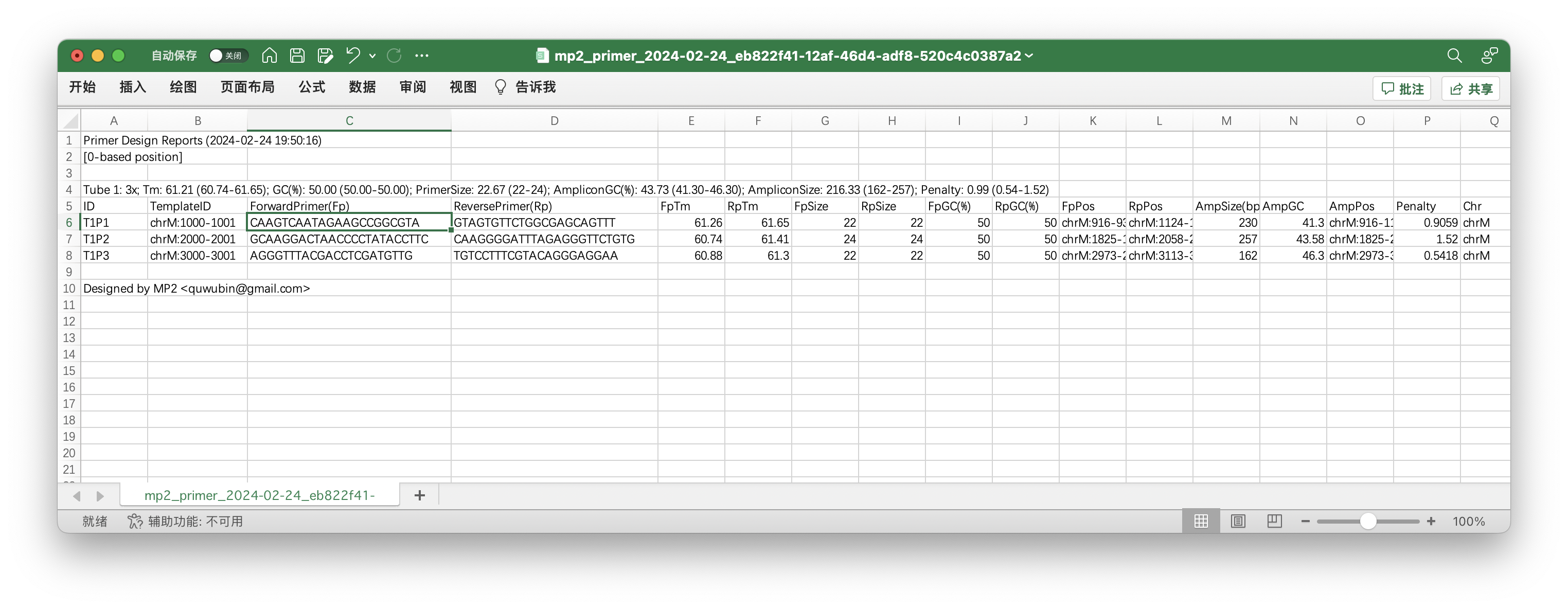 6. Copy the first pair of primers for validation. Paste them on MFEprimer: https://m4.igenetech.com/spec/, and select three databases which we used for designed primers.
6. Copy the first pair of primers for validation. Paste them on MFEprimer: https://m4.igenetech.com/spec/, and select three databases which we used for designed primers.
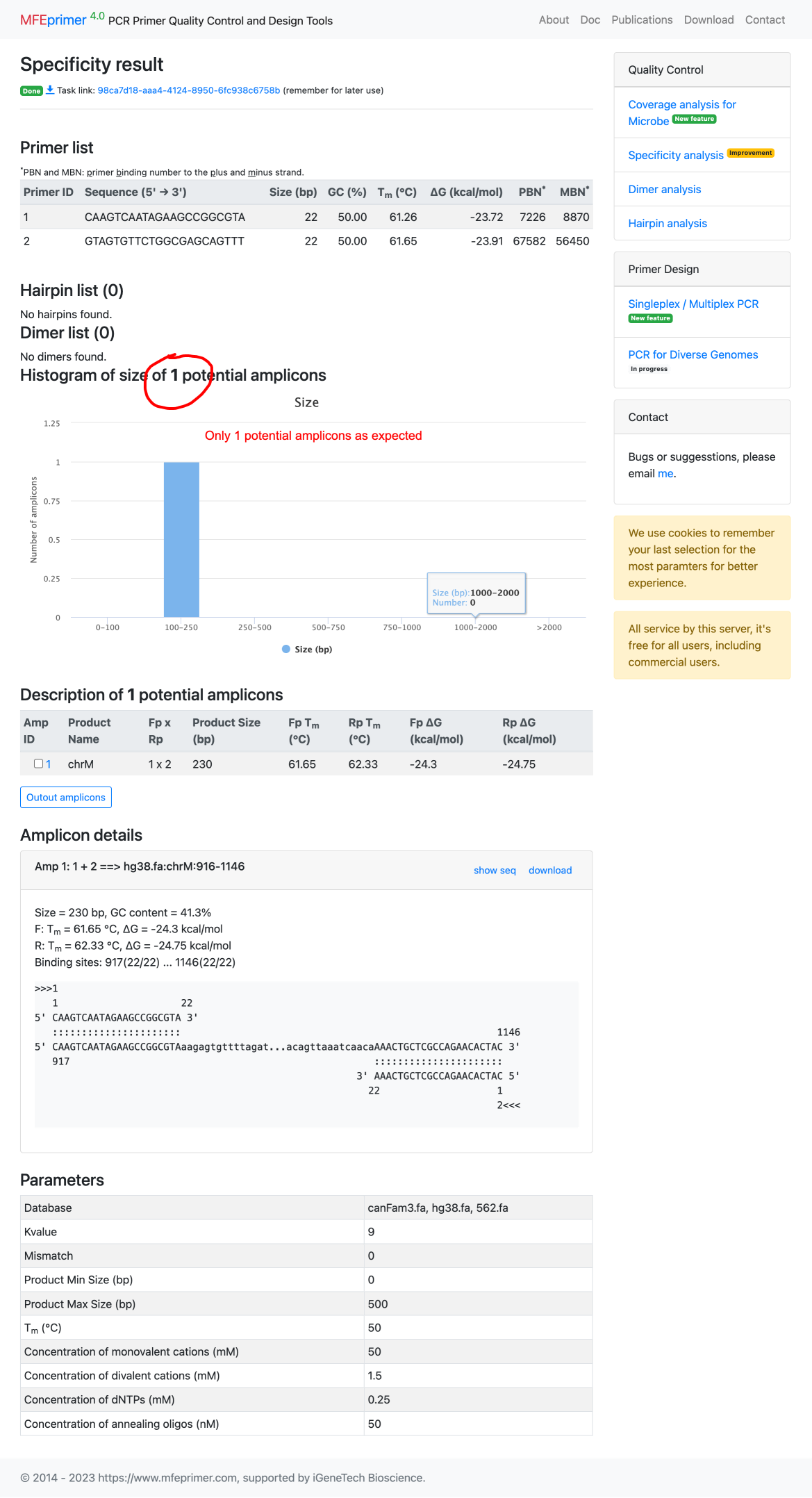
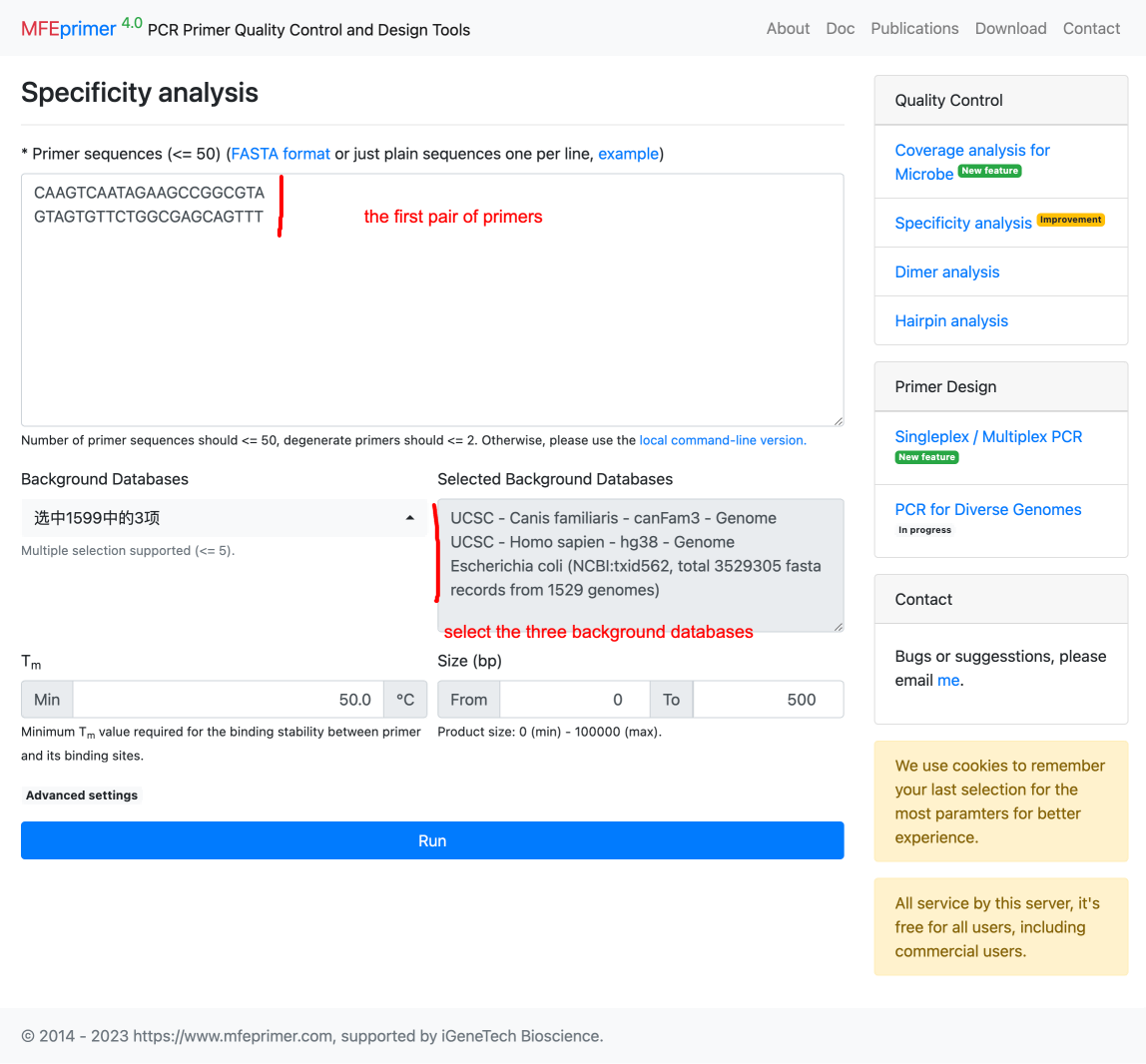 7. MFEprimer shows only one amplicon found as we expected.
7. MFEprimer shows only one amplicon found as we expected.
Designing primers capable of amplifying both genome A and genome B simultaneously #
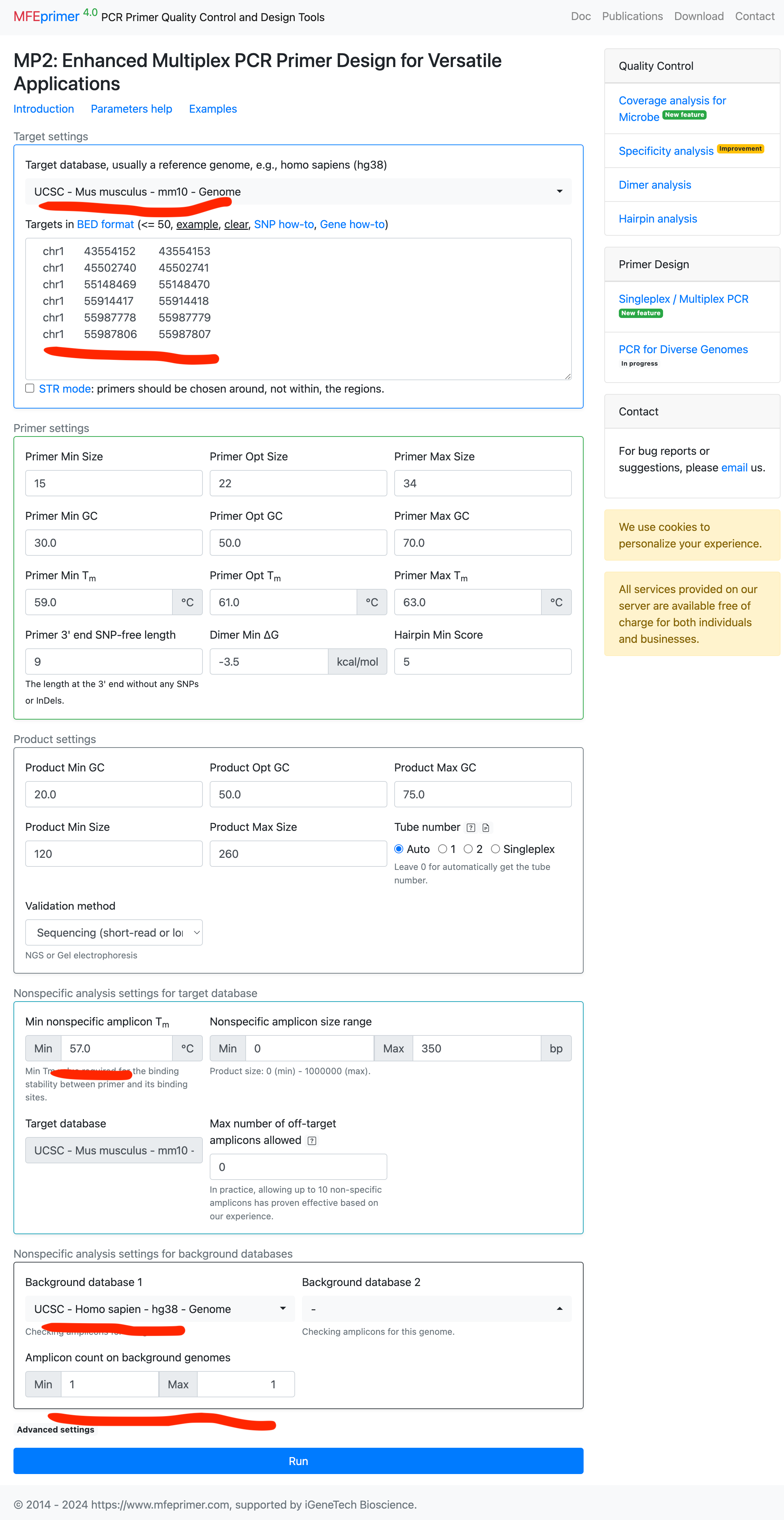
- Select the target database: this time we design priemrs for mouse and requires these primers should also have exactly one amplicon in human genome. Please be noted that we also set “Min nonspecific amplicon Tm” = 57, a bigger higher Tm. This is to tell the server we need a good (with Tm close to the target amplicon in mouse genome) amplicon for human genome.
- Enter out targets in BED format;
chr1 43554152 43554153
chr1 45502740 45502741
chr1 55148469 55148470
chr1 55914417 55914418
chr1 55987778 55987779
chr1 55987806 55987807
- Also select backgrounds databases.
- And set “Nonspecific amplicon count on background genome” = 1.
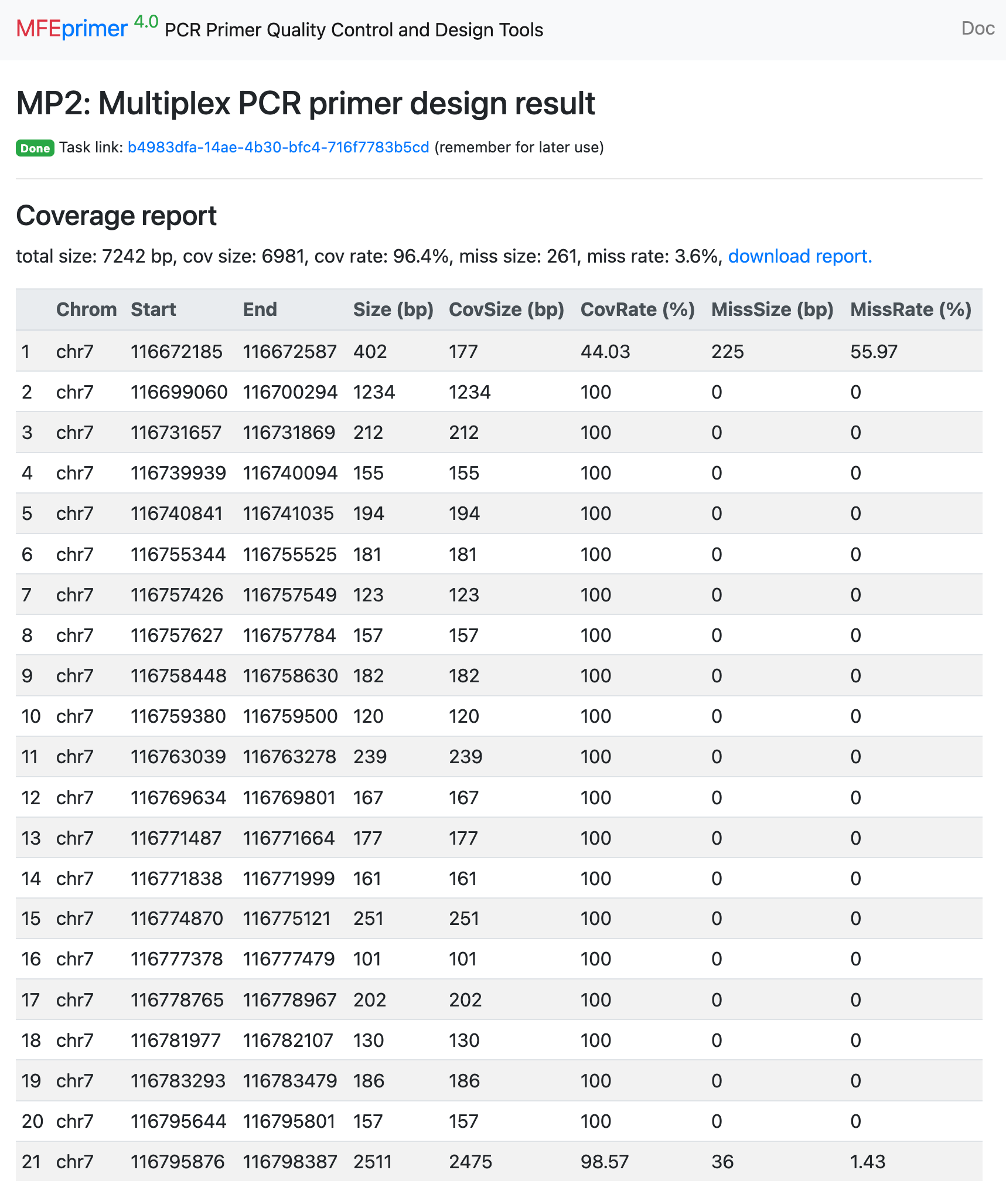
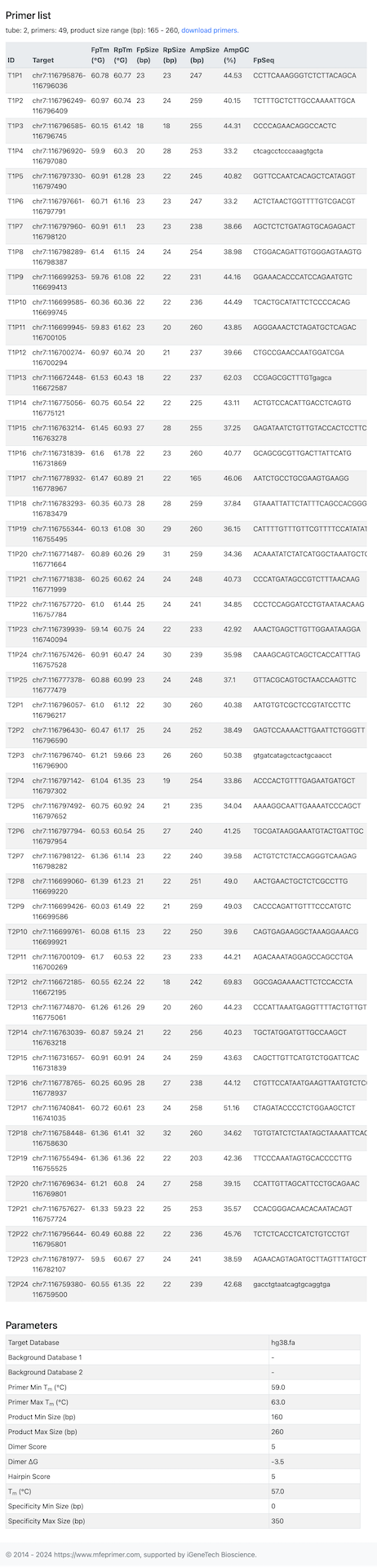
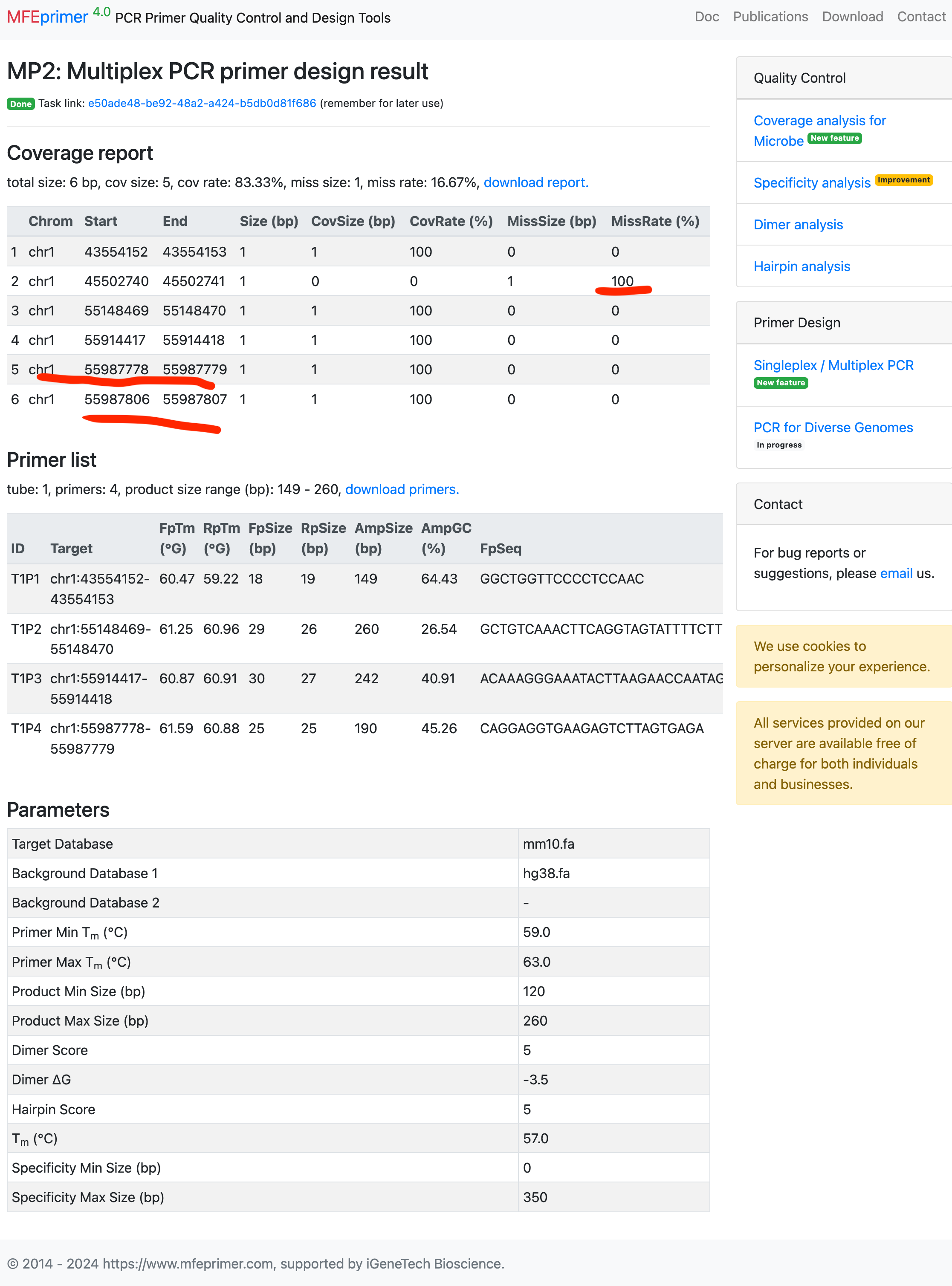
- The result page contains two parts, the first is “Coverage report”, listing all the target coverage in details. The second is “Primer list”, list all the primers in details. Each of them can be downloaded for further use. In this example, only one target can’t find a proper pair of primers to amplify both the mouse and human genome. And to be noted that, the last two targets is close enough that one pair of primers can amplify them both. So ther is only 4 pair of primers can amplify 5 targets.
Designing multiplex primers with different amplicon sizes for capillary electrophoresis #
- Select the target database;
- Enter the targets in BED format;
- Select the “validation method”;
- And set the “Product Min Different” = 50, this parameter requies the size difference of each amplicons should >= 50 bp.
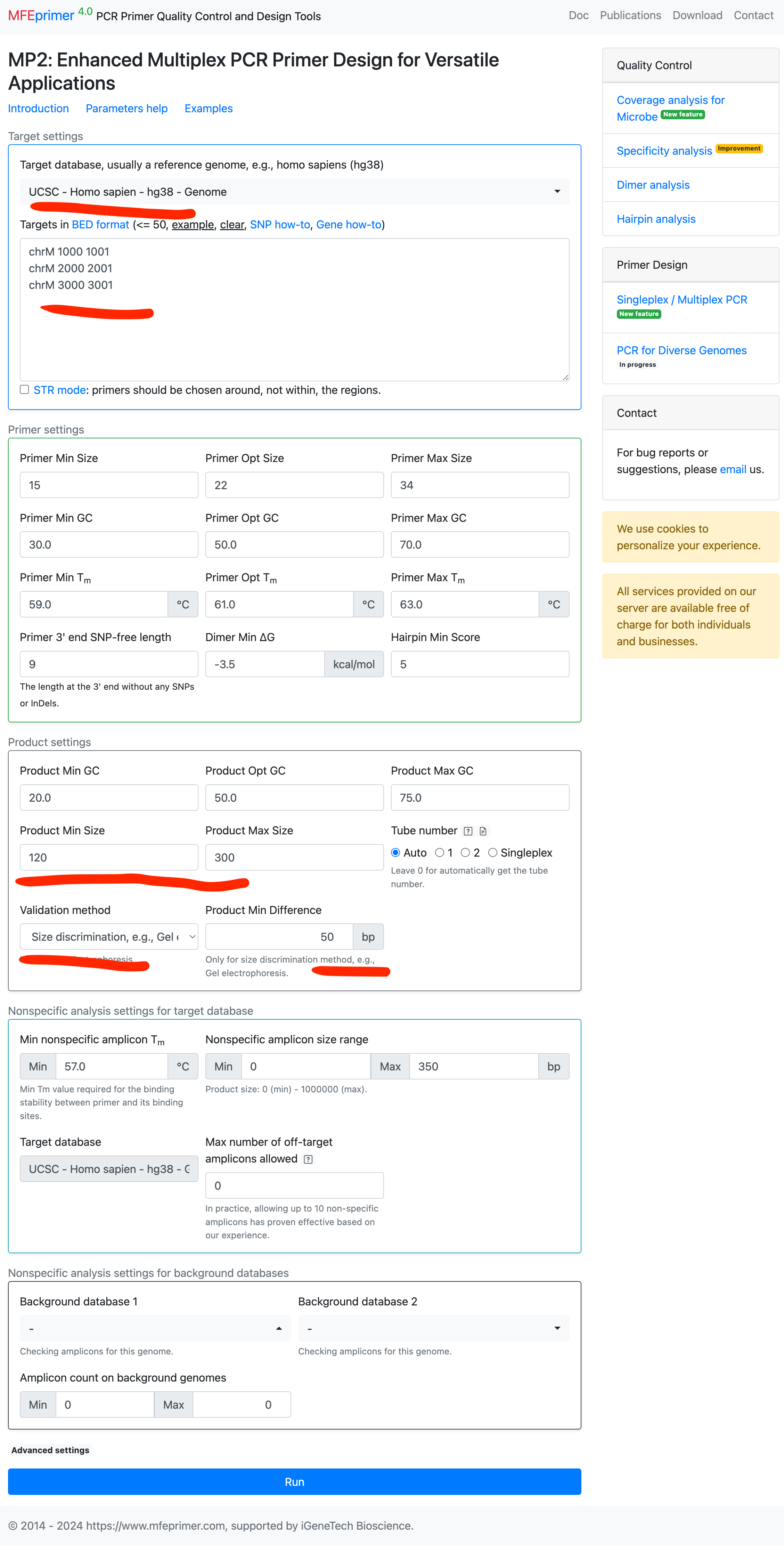
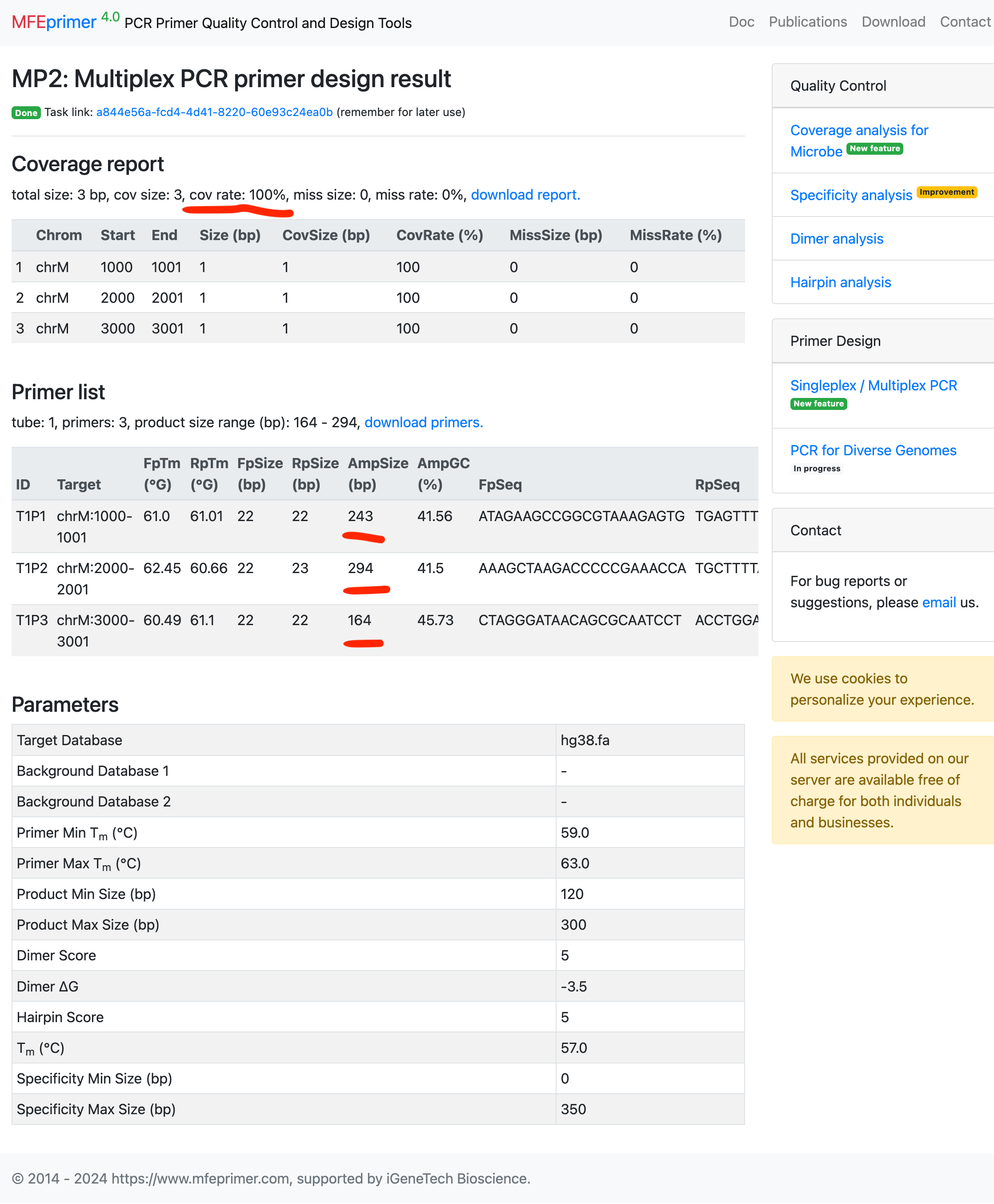
- As expected, the three amplicons have enough size differences which we can discriminate them by capillary electrophoresis.
Designing multiplex primers with similar amplicon sizes for target enrichment, followed by next-generation sequencing. #
- Select the target database and other parameters.
- Enter the targets in BED format, this time we use three gene exons coordinates as targets, as this post did: https://www.mfeprimer.com/posts/gene2bed/
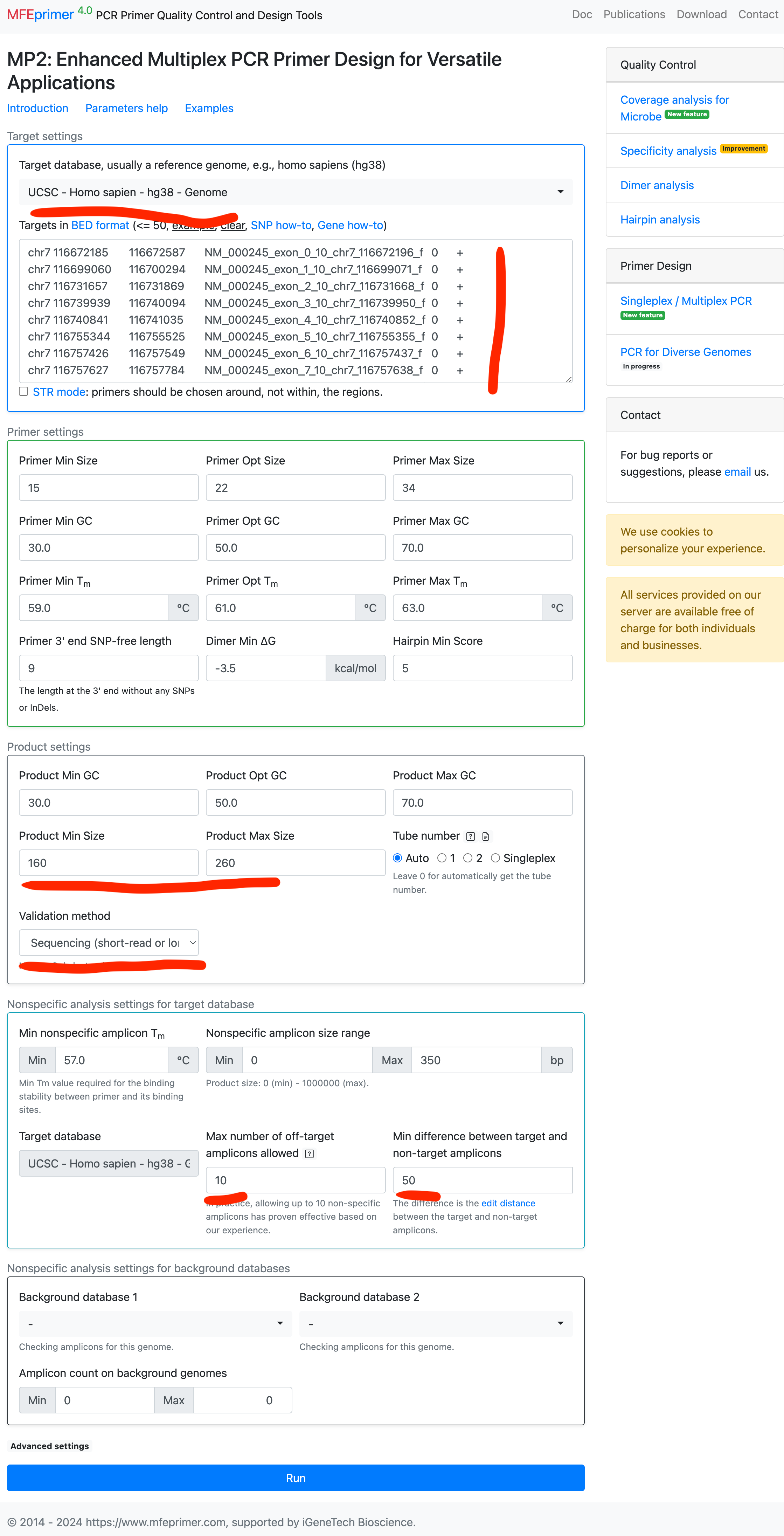
- Here is the result page: https://m4.igenetech.com/mp2/b4983dfa-14ae-4b30-bfc4-716f7783b5cd, and all the amplicons have sizes in range 160-260 bp.